服务热线
欧盟IVDR认证流程的共识文件要点内容
分类:法规更新 发布人:admin 发布时间:1970-01-21 12:58 阅读:929
欧盟体外医疗器械(IVDR)认证流程的共识文件,是制造商向公告机构(NB)申请认证的预申请、申请及申请后阶段例如发放,监控等的流程。
目的:
共识文件旨在详细阐述制造商向公告机构(NBs)申请体外诊断医疗器械(IVD)依据法规 (EU) 2017/746(IVDR)进行认证时,预申请和正式申请的具体流程。文件制定基于对各Team-NB成员现有申请流程及文档的审查,并力求统一这些流程。文件既涵盖依据第110条过渡至IVDR的传统设备,也涉及新上市且未经旧指令认证的设备。此外,文件还简要说明了认证申请完成后的后续认证活动。
适用性:
共识指导文件遵循《体外诊断医疗器械法规》(IVDR) (EU) 2017/746的要求,具体可参考附录VII中关于预申请和申请要求的§4.2、§4.3条款。
不涵盖内容:
IVDR第16条规定的NB证书申请流程。
IVDR附录VII §4.11规定的再认证申请流程。
一般注意事项
共识文件遵循MDCG 2022-14行动16,公告机构制定共同指南协助制造商进行合规评估。为有助于建立共同理解以及减少公告机构收到不完整的认证申请,共识文件提供一个详细的申请过程描述,包括所涉及的步骤以及制造商所需的最少/典型信息和数据。虽协调申请流程中有所需文件的类型和范围,但各公告机构保留要求制造商提供额外信息的权利,相关额外信息应在正式文件中明确并在官网公布。同时共识文件也会随经验积累和法规变化修订。
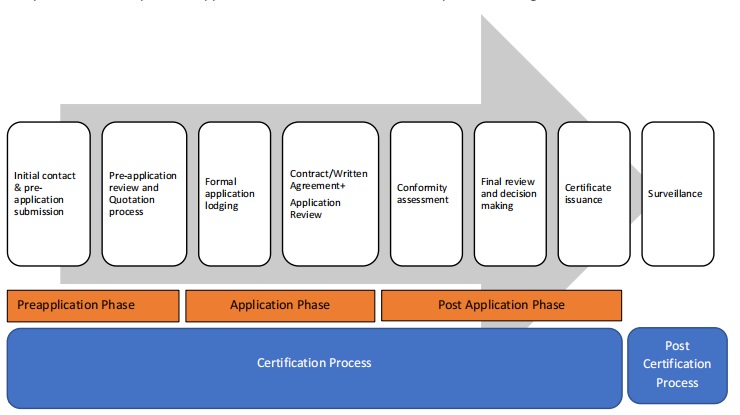
认证流程阶段
(一)初始接触和预申请提交
制造商与公告机构的首次接触,可通过口头或数字方式(如邮件、官网表格)请求合规评估服务。公告机构需审查预申请信息,包括初步验证产品是否在法规范围内及分类,制造商或其欧盟授权代表需提交规定的信息,以便公告机构准备报价,提供详细信息可减少信息补正时间并确保报价准确。
(二)预申请审核和报价流程
公告机构审核制造商提交的预申请信息,初步验证产品是否属IVDR范围及分类是否准确,依据提交的信息(如工厂、分包商 / 供应商、产品等)提供合规评估服务的费用估算,可要求补充信息或澄清以确保报价准确。报价可在后续阶段(如申请审核或合规评估阶段)经制造商同意后修改,部分公告机构可能随报价提供合同模板及条款,其他则在完整提交申请文件后提供。
(三)正式申请提交
-
制造商接受报价后,至少需提交以下信息:
-
质量管理体系评估文件,部分公告机构可能有补充清单。
-
B、C、D 类设备的技术文档
-
自测或近患者检测设备
-
伴随诊断设备(CDx)
-
D 类设备可能需额外文件以符合欧盟参考实验室(EURL)相关实施法规
需提交由制造商或其欧盟授权代表签署的正式申请文件(如表格或信函),旧设备过渡到IVDR 时,无需提交完整技术文档,但需提交技术文档提交计划及设备足够信息供公告机构验证。
(四)合同与申请审核
-
申请提交后,公告机构提供合同文件,双方签署后协议生效,公告机构基于制造商提供的文件进行申请审核。
-
审核内容包括:申请是否符合相关合规评估程序要求、产品是否属器械及分类是否正确、所选合规评估程序是否适用、公告机构是否有能力评估、是否有足够适当资源。
-
审核后决定接受或拒绝申请(仅在合同签署后拒绝),通过EUDAMED或其他方式通知;若制造商撤回申请,公告机构也需通过相应方式通知。
(五)合规性评估
A类设备:
除无菌条件下上市的设备外,无需公告机构合规评估;无菌条件下上市的A类设备,公告机构仅审核与建立、确保和维持无菌条件相关的方面。
B、C、D 类设备:
需结合质量管理体系(QMS)审核、技术文档评估(TDA)和设备测试,可能需要与当局协商等额外程序,D类设备可能需要专家参与评估制造商的性能评估报告(PECP),指定的EURL需验证制造商的性能声明及合规性,公告机构需评估技术文档,必要时涉及多领域专家,出具技术文档评估报告(TDAR)和性能评估报告(PEAR),制造商需处理不符合项,可能需要额外审核。
特殊程序:
D类设备:2024年10月1日后申请的,公告机构需联系EURL进行性能验证;此前申请的,性能验证在更新前进行;不在EURL范围内的,公告机构可自行进行测试。
伴随诊断设备:公告机构需与成员国指定的主管当局或欧洲药品管理局(EMA)协商,提交设备使用说明草案和安全性能摘要(SSP),EMA协商需提前3个月提交 “提交意向书”,根据变更情况决定是否进行新的合规评估或发布补充证书。
最终审核与决策
-
合规评估活动完成后,公告机构进行最终审核和决策,由未参与评估的人员执行,审核内容包括决策报告和支持文件是否完整、是否存在未解决的不符合项。
-
审核结果反映在内部报告中,作为认证过程主要阶段、评估结果的总结,为决策提供建议,决策时考虑最终审核建议、评估文件和其他相关信息,判断是否满足IVDR 要求以决定是否颁发证书。
证书颁发
若决策通过,公告机构按合规途径生成证书,包含IVDR附件XII规定的信息,证书在EUDAMED系统功能完备后上传。
监督活动
-
初始认证后,公告机构确定维持证书所需的监督活动,包括年度QMS审核(在制造商及相关分包商 / 供应商处)、PSUR评估、SSP验证、B和C类设备技术文档抽样评估、警戒数据评估和突击审核。
-
QMS 监督审核至少每年一次,确保制造商维持认证的QMS;技术文档抽样计划按 MDCG 2019-13制定;突击审核至少每五年一次;制造商需提交设备警戒报告,公告机构评估后采取相应行动;D和C类设备需按规定频率提交PSUR 和SSP,SSP 由公告机构验证并在EUDAMED注册;制造商计划对QMS或设备进行重大变更时需通知公告机构,公告机构评估变更影响,决定是否进行额外合规评估活动。
语言要求
技术文档和质量体系文档的语言要求由公告机构指定。

