服务热线




欧盟IVDR Article 2(32)条
'conformity assessment’ means the process demonstrating whether the requirements of this Regulation relatingto a device have been fulfilled:
“符合性评估”是指证明与一个器械有关的本法规(IVDR)的要求是否满足的过程。
其目的是保障体外诊断医疗器械在欧盟市场的安全性和有效性,确保这些器械能够准确、可靠地用于疾病诊断、监测、预防等医疗目的,保护患者和使用者的健康与安全。同时建立统一的评估标准和流程,促进欧盟内部体外诊断医疗器械市场的一体化,消除贸易壁垒,使符合要求的产品能够在各成员国之间自由流通。
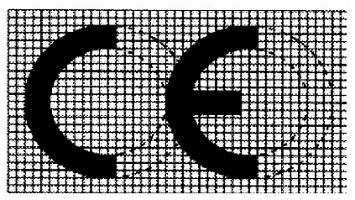
ANNEX V要求符合性评估的产品上市后需要加贴合格的CE标志。CE标志应由 “CE” 字样组成,采用以下样式:
Article 48 Conformity assessment procedures(第4符合性评估程序)
介绍了各类别产品的符合性评估程序的要求
ANNEX IX
CONFORMITY ASSESSMENT BASED ON A QUALITY MANAGEMENT SYSTEM AND ON ASSESSMENT OF TECHNICAL DOCUMENTATION
基于质量管理体系和技术文件评估的符合性评估
ANNEX X
CONFORMITY ASSESSMENT BASED ON TYPE-EXAMINATION
基于型式检验的符合性评估
ANNEX XI
CONFORMITY ASSESSMENT BASED ON PRODUCTION QUALITY ASSURANCE
基于生产质量保证的符合性评估
IVDR Article 48
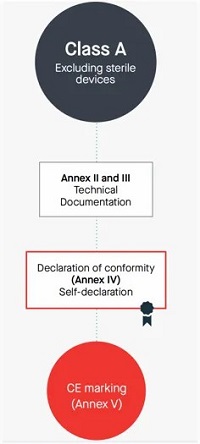
10.Manufacturers of class A devices, other than devices for performance study, shall declare the conformity of their products by issuing the EU declaration of conformity referred to in Article 17, after drawing up the technical documentation set out in Annexes II and III.
Class A 非无菌产品
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
Self-declaration,不涉及公告机构审核和发证
IVDR Article 48
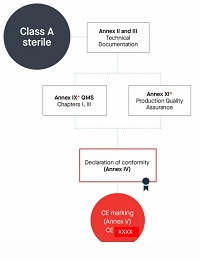
10.Manufacturers of class A devices, other than devices for performance study, shall declare the conformity of their products by issuing the EU declaration of conformity referred to in Article 17, after drawing up the technical documentation set out in Annexes II and III.
However, if those devices are placed on the market in sterile condition, the manufacturer shall apply the procedures set out in Annex IX or in Annex XI. Involvement of the notified body shall be limited to the aspects relating to establishing, securing and maintaining sterile conditions.
Class A 无菌产品
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合AnnexIX 质量管理体系,或者Annex Xl生产质量保证
但仅限无菌方面(公告机构的参与应限于与建立、确保和维持无菌状态相关的方面。)
IVDR Article 48
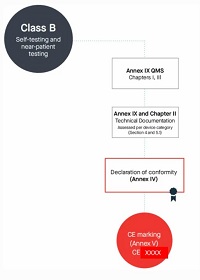
9. Manufacturers of class B devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I and III ►C1 of Annex IX, and, in addition, to an assessment of the technical documentation as specified in Section 4 of that Annex for at least one representative ◄ device per category of devices.
Class B(自测和床旁检测除外)
按照Annex lX第一章和第三章进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个器械类别至少一个代表器械进行技术文档的评估,依据Annex lX的Section 4

IVDR Article 48
9.Manufacturers of class B devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I and III ►C1 of Annex IX, and, in addition, to an assessment of the technical documentation as specified in Section 4 of that Annex for at least one representative ◄ device per category of devices.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for assessment of the technical documentation set out in Section 5.1 of Annex IX.
Class B自测和床旁检测产品
按照Annex lX第一章和第三章进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个器械类别至少一个代表器械进行技术文档的评估,依据Annex lX的Section 4 和5.1
IVDR Article 48
7. Manufacturers of class C devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I and III ►C1 of Annex IX, and, in addition, to an assessment of the technical documentation as specified in Section 4 of that Annex for at least one ◄ representative device per generic device group.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
Class C(自测、床旁检测、伴随诊断除外)
按照Annex lX第一章和第三章进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个通用器械组的至少一个代表器械进行技术文档的评估依据Annex IX的Section 4

IVDR Article 48
7.Manufacturers of class C devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I and III ►C1 of Annex IX, and, in addition, to an assessment of the technical documentation as specified in Section 4 of that Annex for at least one ◄ representative device per generic device group.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
Class C自测和床旁检测产品
按照Annex lX第一章和第三章进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个通用器械组的至少一个代表器械进行技术文档的评估依据Annex IX的Section 4和Section 5.1

IVDR Article 48
7. Manufacturers of class C devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I and III ►C1 of Annex IX, and, in addition, to an assessment of the technical documentation as specified in Section 4 of that Annex for at least one ◄ representative device per generic device group.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
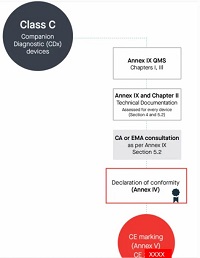
In addition to the procedures referred to in the first and second subparagraphs, for companion diagnostics the notified body shall for every device follow the procedure for technical documentation assessment laid down in Section 5.2 of Annex IX, and shall apply the procedure for technical documentation assessment laid down in Sections 4.1 to 4.8 of Annex IX and shall consult the competent authority designated by the Member States in accordance with Directive 2001/83/EC or the EMA, as applicable, in accordance with the procedure set out in Section 5.2 of Annex IX.
Class C伴随诊断产品
按照Annex lX第一章和第三章进行进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
针对每个器械进行技术文档的评估,依据Annex IX的Section 4、5.1、5.2
咨询成员国指定的主管当局或者欧洲药品管理局。
8. Manufacturers of class C devices, other than devices for performance study, may, instead of the conformity assessment procedure pursuant to paragraph 7, choose to apply a conformity assessment as specified in Annex X coupled with a conformity assessment as specified in Annex XI except its Section 5.
For companion diagnostics the notified body shall in particular for every device consult a competent authority designated by the Member States in accordance with Directive 2001/83/EC or the EMA, as applicable, in accordance with the procedure set out in point (k) of Section 3 of Annex X.
Class C除Annex lX质量管理体系+技术文档的路径外,还可以:
(即:不采用上面的第7条中的评定程序)
Annex X基于型式检验的符合性评估+Annex Xl 基于生产质量保证的符合性评估(section 5的class D Verification除外)
属于伴随诊断的产品,还需要咨询主管当局或EMA
IVDR Article 48
3.Manufacturers of class D devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I, II except for Section 5, and in Chapter III of Annex IX.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
5. In particular, and without prejudice to any of the obligations pursuant to the other procedures referred to in paragraphs 3 and 4, for devices for which one or more EU reference laboratories have been designated in accordance with Article 100, the notified body performing the conformity assessment shall request one of the EU reference laboratories to verify by laboratory testing the performance claimed by the manufacturer and the compliance of the device with the applicable CS, or with other solutions chosen by the manufacturer to ensure a level of safety and performance that is at least equivalent, as specified in Section 4.9 of Annex IX and in point (j) of Section 3 of Annex X. Laboratory tests performed by an EU reference laboratory shall in particular focus on analytical and diagnostic sensitivity using the best available reference materials.
Class D(有CS)(伴随诊断除外)产品符合性评估路径
按照Annex lX第一章,第二章(第5节除外),第三章进行进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
Annex lX质量管理体系
每个器械进行技术文档的评估,依据AnnexIX的Section 4、5.1
当器械有1个或多个参考实验室指定时,公告机构需要要求至少一个EU参考实验室对于制造商宣称的性能及CS的合规性进行验证,特别是分析和诊断灵敏度。

IVDR Article 48
3.Manufacturers of class D devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I, II except for Section 5, and in Chapter III of Annex IX.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
6. In addition to the procedure applicable pursuant to paragraphs 3 and 4, where no CS are available for class D devices and where it is also the first certification for that type of device, the notified body shall consult the relevant experts referred to in Article 106 of Regulation (EU) 2017/745 on the performance evaluation report of the manufacturer. To that end, the notified body shall provide the performance evaluation report of the manufacturer to the expert panel within five days of receiving it from the manufacturer. The relevant experts shall, under the supervision of the Commission, provide their views, in accordance with Section 4.9 of Annex IX or point (j) of Section 3 of Annex X, as applicable, to the notified body within the deadline for delivery of the scientific opinion by the EU reference laboratory as specified therein.
Class D(无CS)(伴随诊断除外)产品符合性评估路径
按照Annex lX第一章,第二章(第5节除外),第三章进行进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个器械进行技术文档的评估,依据Annex IX的Section4、5.1
当器械有1个或多个参考实验室指定时,公告机构需要要求至少一个EU参考实验室对于制造商宣称的性能进行验证,特别是分析和诊断灵敏度。
没有CS,且为该类型的首次认证,公告机构要就制造商的性能评估报告咨询 (EU)2017/745法规第106条提及的相关专家,在收到报告5天内提供给专家小组,专家要在欧盟参考实验室给出科学意见的截止日期内,根据附件IX第4.9节或附件X第3节(j)点给出意见。

IVDR Article 48
3.Manufacturers of class D devices, other than devices for performance study, shall be subject to a conformity assessment as specified in Chapters I, II except for Section 5, and in Chapter III of Annex IX.
In addition to the procedures referred to in the first subparagraph, for devices for self-testing and near-patient testing, the manufacturer shall follow the procedure for technical documentation assessment set out in Section 5.1 of Annex IX.
Class D伴随诊断产品符合性评估路径
按照Annex lX第一章,第二章(第5节除外),第三章进行合规评定
编写Annex Il技术文档+Annex Ⅲ上市后监督技术文档
符合Annex lX质量管理体系
每个器械进行技术文档的评估,依据AnnexIX的Section 4、依据Annex lX的Section 4、5.1、5.2
咨询成员国指定的主管当局或者欧洲药品管理局。(Annex lX, Section 5.2)
当器械有1个或多个参考实验室指定时,公告机构需要要求至少一个EU参考实验室对于制造商宣称的性能进行验证,特别是分析和诊断灵敏度。
没有CS,且为该类型的首次认证,公告机构要就制造商的性能评估报告咨询 (EU)2017/745法规第106条提及的相关专家,在收到报告5天内提供给专家小组,专家要在欧盟参考实验室给出科学意见的截止日期内,根据附件IX第4.9节或附件X第3节(j)点给出意见。
4. Manufacturers of class D devices, other than devices for performance study, may, instead of the conformity assessment procedure applicable pursuant to paragraph 3, choose to apply a conformity assessment as specified in Annex X coupled with a conformity assessment as specified in Annex XI.
For companion diagnostics, the notified body shall in particular consult a competent authority designated by the Member States in accordance with Directive 2001/83/EC or the EMA, as applicable, in accordance with the procedure set out in point (k) of Section 3 of Annex X.
Class D除Annex lX质量管理体系+技术文档的路径外,还可以:
(即:不采用上面的第3条中的评定程序)
Annex X基于型式检验的符合性评估+Annex Xl 基于生产质量保证的符合性评估
属于伴随诊断的产品,还需要咨询主管当局或EMA


