服务热线
欧盟 IVDR 设备分类规则要点梳理
分类:法规更新 发布人:admin 发布时间:2025-01-02 00:00 阅读:657
欧盟《体外诊断医疗器械法规》(In Vitro Diagnostic Medical Device Regulation, IVDR,EU 2017/746)引入了新的分类规则,将体外诊断医疗器械(IVD)划分为A类、B类、C类和D类,以风险为基础进行分级。以下是IVDR分类规则的核心内容:
1. 分类依据
IVDR使用的是基于风险的分类体系,主要考虑以下因素:
医疗器械的预期用途。
检测的临床目标(如是否检测高风险感染性病原体、遗传信息等)。
使用人群(普通患者、孕妇、新生儿等)的特殊性。
检测结果对个人或公共健康的潜在影响。
2. 分类规则
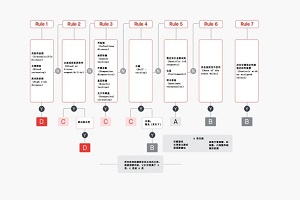
根据IVDR附件VIII,IVD被划分为以下四类:
A 类(低风险):
此类器械仅用于普通实验室用途,不会与患者样本发生直接接触,例如常见的培养基等产品。该类器械对个人或公共健康产生的影响较为微小,在整个医疗器械风险层级中处于较低水平。
B 类(较低风险):
涵盖了众多常规的体外诊断医疗器械,在分类规则中,那些未被明确划定为 C 类或 D 类的器械通常会被归入此类。像部分常见的生化检测设备即属于这一类别。使用 B 类器械对个人而言存在中等程度的风险,不过其对公共健康的影响相对较小。
C 类(较高风险):
使用 C 类器械预期会使个人面临较高的风险,对公共健康也存在中等程度的风险影响。例如能够检测高风险病原体(如乙型肝炎病毒)的设备,适用于遗传检测以及癌症标志物检测的器械,还有用于特定高风险用途的人类遗传材料检测的相关设备等都属于这一类。
D 类(高风险):
这类器械对个人或公共健康具有最为显著的影响,属于风险层级中的最高级别。比如用于检测高度传染性病原体(如 HIV、HCV)的设备,以及供血、供器官或细胞治疗的相关筛查设备等。这些器械往往涉及到公共健康危害或者生命安全风险等关键问题。
3. 分类的重要性
合规性要求:
不同分类要求不同程度的合规流程,高风险类别(C类和D类)需要更严格的评估程序,包括第三方认证机构的审核。
公告机构介入:
根据风险等级,IVDR引入公告机构(Notified Body)参与大部分IVD的认证过程,特别是C类和D类设备。
为了更深入地明晰 IVDR 的分类规则,接下来将对其进行系统梳理与详细阐释
设备用于以下目的归为D类
用于检测血液、血液成分、细胞、组织或器官或其任何衍生物中是否存在或接触传染性病原体的设备,以评估其是否适合输血、移植或细胞给药。
用于检测是否存在或暴露于可导致危及生命的疾病的传播媒介的设备,该疾病具有高或疑似高传播风险。
用于确定危及生命的疾病的感染负荷的设备,其中监测在患者管理过程中至关重要。
Rule1 Class D的例子:
艾滋病病毒
乙型肝炎病毒
出血热病毒
新型冠状病毒等
用于血型鉴定或确定胎儿与母体血型不相容性的设备,或用于确保输血、移植或细胞给药的血液、血液成分、细胞、组织或器官的免疫相容性的组织分型设备,被归类为C类
除非旨在确定以下任何标记:
–ABO血型系统
–恒河猴系统
–Kell系统
–基德系统
–达菲系统,在这种情况下,它们被归类为D类
常规血液或组织兼容为C类
高危血型为D类
以下情况设备将被归类为C类。
a、用于检测性传播媒介存在或暴露的设备
b、用于检测脑脊液或血液中是否存在感染源而不存在高传播风险或疑似高传播风险的设备
c、用于检测传染源存在的设备,如果错误的结果有可能导致被检测的个人、胎儿或胚胎或其后代死亡或严重残疾
d、用于对妇女进行产前筛查以确定其对传染性病原体的免疫状态的设备
e、用于确定感染性疾病状态或免疫状态的设备,其中存在错误结果导致患者管理决策的风险,从而导致患者或患者后代的生命危险
f、拟用作伴随诊断的设备
g、用于疾病分期的设备,存在错误结果导致患者管理决策的风险,从而导致患者或患者后代的生命危险
h、用于癌症筛查、诊断或分期的设备
i、用于人类基因检测的设备
j、用于监测药品、物质或生物成分水平的设备,当存在错误结果可能导致患者管理决策,从而对患者或患者后代造成危及生命的情况时
k、用于管理患有危及生命的疾病或病症的患者的设备
l、用于筛查胚胎或胎儿先天性疾病的设备
m、用于筛查新生儿先天性疾病的设备,如果不能检测和治疗这些疾病,可能会导致危及生命的情况或严重残疾
属于rule 3的传染病、癌症检测、伴随诊断、基因检测、先天性筛查,为Class C
Rule 3 Class c的例子:
Human papilloma virus (HPV)
(a)用于自我检测的设备被归类为C类,但用于检测怀孕、生育能力测试和测定胆固醇水平的设备以及用于检测尿液中葡萄糖、红细胞、白细胞和细菌的设备除外,这些设备被归为B类
(b)用于床旁检测的设备本身就被分类了
自测为Class C,除特殊的Class B外
床旁检测可以为Class B、C、D,依据其产品特性及预期用途
以下设备被归类为A类:
a、制造商为使其适用于特定检查的体外诊断程序而设计的一般实验室用产品、不具有关键特性的配件、缓冲溶液、洗涤溶液、一般培养基和组织学染色剂
b、制造商专门用于体外诊断程序的仪器。规则5b适用于制造商专门用于体外诊断程序的仪器。这些仪器被归类为A类,而试剂和试剂盒则单独分类。
c、样本容器。样本容器是指用于体外诊断检查的生物样本(如细胞、组织样本、尿液、粪便)的主要容纳、保存、运输和储存的样本容器或抽真空或非抽真空管,这些容器或管是空的或预充有固定剂溶液或其他通用试剂的。它们被归为A类。
一般实验室使用的产品、仪器、样品容器,为Class A
上述分类规则未涵盖的设备被归类为B类.规则6适用于规则1-5未涵盖的设备。
没有规则可以靠的,按照rule 6为class B
没有定量或定性赋值的质控,为Class B
根据ANNEX VIII 1.6 有定量或定性赋值的质控,与配合使用的器械为相同分类
本法规不适用于以下情况
-
仅供一般实验室使用的产品或仅供研究用途的产品,除非此类产品鉴于其特性被制造商明确指定用于体外诊断检查;
-
侵入性取样产品或为获取样本而直接应用于人体的产品;
-
国际认证的参考物质;
-
用于外部质量评估方案的材料。
依据Article 1的3(C)和(d),国际认证参考材料,外部质评材料,不属于IDR监管的范围。