服务热线
法规注册咨询
分类:法规注册咨询 发布人:admin 发布时间:2024-09-06 15:50 阅读:2869
全球法规注册咨询服务
国瑞中安专注全球法规注册咨询服务,包括中国NMPA,俄罗斯RZN,欧盟CE MDR&IVDR,美国FDA&510K,澳洲TGA,巴西ANVISA,韩国MFDS,加拿大MDL&MDEL,英国MHRA、UKCA,欧亚经济联盟EAEU,东南亚注册,中东注册,独联体国家注册等
医疗器械NMPA注册
医疗器械NMPA注册是指在中国国家药品监督管理局(NMPA,National Medical Products Administration)进行注册和审批的过程,以获得在中国市场上销售和使用的合法资格。NMPA是中国的药品监管机构,负责监督和管理药品、医疗器械、化妆品等产品的安全性和质量。国内医疗器械的NMPA注册是指根据中国国家药品监督管理局(NMPA)的要求,将医疗器械产品纳入中国市场并进行注册的过程。
中国如何定义医疗器械?
《医疗器械监督管理条例》第八章附则中医疗器械定义是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。
医疗器械的使用旨在达到下列预期目的:
1.对疾病的预防、诊断、治疗、监护、缓解。
2.对损伤或者残疾的诊断、治疗、监护、缓解、补偿。
3.对解剖或者生理过程的研究、替代、调节或者支持。
4.对生命的支持或者维持。
5.妊娠控制。
6.通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
中国将医疗器械按照不同风险分为三类:
· 第一类医疗器械(低风险)如:纱布、绷带、医用退烧贴等
· 第二类医疗器械(中风险)如,输液器、手术手套、理疗仪器等
· 第三类医疗器械(高风险),如心脏支架、心脏起搏器、人工关节等
第一类医疗器械实行产品备案管理。第二类、第三类医疗器械实行产品注册管理。
· 境内第一类医疗器械备案,备案人向设区的市级负责药品监督管理的部门提交备案资料。
· 境内第二类医疗器械由省、自治区、直辖市药品监督管理部门审查,批准后发给医疗器械注册证。
· 境内第三类医疗器械由国家药品监督管理局审查,批准后发给医疗器械注册证。
· 进口第一类医疗器械备案,备案人向国家药品监督管理局提交备案资料。
进口第二类、第三类医疗器械由国家药品监督管理局审查,批准后发给医疗器械注册证。
国瑞中安为医疗器械企业提供如下全流程咨询辅导注册服务:

美国医疗器械FDA认证
医疗器械的FDA认证是指通过美国食品药品监督管理局(FDA)的审批程序,获得在美国市场上销售和使用的合法授权。这是医疗器械制造商必须遵循的重要法规,以确保其产品在安全和有效性方面符合美国的监管标准。
FDA对医疗器械的监管控制及分类取决于器械的预期用途,使用说明以及风险程度。选择正确的监管递交路径,以确保器械的安全性和有效性。
FDA对每种医疗器械明确规定了其产品分类和管理要求,按照不同风险等级分成三类:
Ⅰ类-低等风险(一般控制), 例如:牙刷、
Ⅱ类-中等风险(一般控制以及特殊控制),例如:无创血压监测器
Ⅲ类-高等风险(一般控制以及上市前批准),例如:心脏瓣膜
按照递交注册路径分为五种:
1. 510K(上市前通知):适用于一些中高风险医疗器械的市场准入途径,找到已获FDA批准注册并上市的同类产品,并证明自家产品与该同类产品具有相同或相似的安全性和有效性。
2. 豁免510K:大部分Ⅰ类和小部分Ⅱ类产品由于风险等级较低且产品技术成熟,可以理解需要在FDA官网注册,但不需经过FDA审批便可上市销售。
3. De Novo(分类请求):称为创新型器械的注册途径,风险等级判定为I类或II类,且无法找到同类产品完成510k注册途径。
4. PMA(上市前批准):医疗器械FDA上市申报方式中最为严格的制度,适用于大部分III类需要采用的途径,如人工心脏瓣膜、心脏起搏器等。
5. HDE (Humanitarian Device Exemption):罕见病所用到的器械采取的途径,比如:治疗多发性骨髓瘤患者用的人造肾脏。
国瑞中安提供医疗器械FDA注册流程:
1. 确定器械所属分类:Ⅰ类、Ⅱ类或Ⅲ类
2. 选择正确的上市前注册路径:510k、豁免510k、De novo、PMA、HDE
3. 准备上市前注册所需的信息,主要包括以下四个方面:
(1)设计控制:所有Class II和Class III器械必须按照质量体系法规(21 CFR 820.30)的设计控制进行设计。
(2)非临床测试:基于器械的操作性能、材料等在GLP实验室检测。
(3)临床证据:PMA, HDE、一些510(k)和De Novo需要临床证据。
(4)标识:FFDCA Section 201(k) 定义的标识包括标签、宣传册、用户手册、说明书等
4、递交注册信息至FDA审查
5、提交资料审查之前,需先完成FDA官方缴费
6、FDA在收到资料并确认企业已完成缴费,便会对资料后进行审查
7、审查通过后,进行企业注册和产品列名后便可上市销售
Notice:记得准时向FDA缴纳注册年金,否则产品下一自然年不能上市售卖哦。
欧盟医疗器械CE认证
自2021年5月起,欧盟医疗器械新法规2017/745(MDR法规)正式实施,意味着医疗器械应满足MDR法规并获得CE标志批准后,方可在欧洲经济区的31个国家/地区销售设备。
CE-MDR将医疗器械分为4个风险等级:
I类(低风险),如:手动轮椅、听诊器、绷带
IIa类(中低风险),如:正畸线、无创无呼吸、血氧仪、
IIb类(中高风险),如:人工晶状体、婴儿保温箱
III类(高风险),如:起搏器、人工心脏瓣膜、心血管缝合线
31个国家/地区分别是:
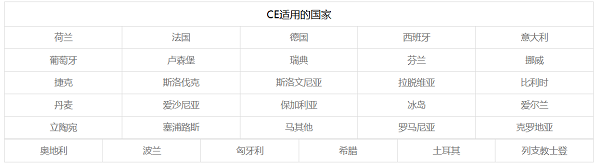
Notice:除美国、日本、澳大利亚等国,其它绝大多数国家认可CE认证转换的自由销售证书CFS/FSC。
医疗器械CE认证流程图:
.png "ce流程(1).png")
欧盟体外诊断ce认证
众所周知,自测CE认证最大的困难点就在于如何实施与完成临床试验研究及可用性研究。
国瑞中安能高效制定出一版有效、靠谱的可行性方案,确保完成后的临床报告与可用性研究报告能得到公告机构的百分百认可。
其次,公告机构对于技术文档的审核要求很高,具体需要其涵盖内容完整规范,必须符合法规要求。对于企业来说,撰写这样一份涵盖内容广泛且跨应用领域的技术文档,其难度是非常大的。
国瑞中安拥有丰富的欧洲市场准入经验,能根据客户产品和欧盟最新法规,编写出完善、合规的技术文档,为企业节约大量的时间和金钱。
国瑞中安可为客户提供以下帮助:从案件的申请受理到技术文件的准备、预审核,临床实验研究解决方案、可用性研究解决方案、审核整改协助、获得证书,提供全过程、全方位的技术咨询服务,和渠道资源支持,确保合作企业能在第一时间成功完成注册。
国瑞中安能高效制定出一版有效、靠谱的可行性方案,确保完成后的临床报告与可用性研究报告能得到公告机构的百分百认可。
其次,公告机构对于技术文档的审核要求很高,具体需要其涵盖内容完整规范,必须符合法规要求。对于企业来说,撰写这样一份涵盖内容广泛且跨应用领域的技术文档,其难度是非常大的。
国瑞中安拥有丰富的欧洲市场准入经验,能根据客户产品和欧盟最新法规,编写出完善、合规的技术文档,为企业节约大量的时间和金钱。
国瑞中安可为客户提供以下帮助:从案件的申请受理到技术文件的准备、预审核,临床实验研究解决方案、可用性研究解决方案、审核整改协助、获得证书,提供全过程、全方位的技术咨询服务,和渠道资源支持,确保合作企业能在第一时间成功完成注册。
截至目前,国瑞中安累计帮助国内外各大IVD企业斩获几十张自测CE认证证书,累计为超100家医疗器械企业提供了专业的注册咨询服务,其中不乏有诸多头部企业的支持和青睐,如:圣湘生物、奥泰生物、博拓生物、安旭生物、基蛋生物、九强生物、康泰医学、安图生物、华大基因、亚辉龙、宝太生物、微策生物、微创医疗等知名企业,在市场中独占鳌头。
值得一提的是,国瑞中安协助企业完成的所有临床性能评估及Laymen可用性研究均得到了公告机构的完全认可!实现了100%的成功率,无一失败案例!
(1).png "国瑞中安承接新冠自测CE认证.png")
部分IVD企业证书截图
众所周知,体外诊断产品CE认证最大的困难点就在于如何实施与完成临床实验研究及可用性研究。
国瑞中安能高效制定出一版有效、靠谱的可行性方案,确保完成后的临床报告与可用性研究报告能得到公告机构的百分百认可。
其次,公告机构对于技术文档的审核要求很高,具体需要其涵盖内容完整规范,必须符合法规要求。对于企业来说,撰写这样一份涵盖内容广泛且跨应用领域的技术文档,其难度是非常大的。
国瑞中安拥有丰富的欧洲市场准入经验,能根据客户产品和欧盟最新法规,编写出完善、合规的技术文档,为企业节约大量的时间和金钱。
为什么选择国瑞中安?
1.合作经验、资源丰富
国瑞中安在体外诊断产品国际注册及临床研究解决方案方面积累了大量的成功经验和资源,目前国瑞中安已与国外多家临床实验机构建立了良好的合作关系。
2.全流程、全方位服务
国瑞中安可为客户提供以下帮助:从案件的申请受理到技术文件的准备、预审核,临床实验研究解决方案、可用性研究解决方案、审核整改协助、获得证书,提供全过程、全方位的技术咨询服务,和渠道资源支持,确保合作企业能在第一时间成功完成注册。
3.团队高效、获证迅速
有合作需求的企业,可联系国瑞中安!
医疗器械认证相关疑问,欢迎您垂询:17715243573

